蛋白质相互作用研究能够从分子水平上揭示蛋白质的功能,帮助揭示生长发育、新陈代谢、分化和凋亡等细胞活动的规律。在全基因组范围内识别蛋白质相互作用对是解释细胞调控机制的重要一步。随着蛋白质相互作用实验技术的发展,人们能够获得大量的蛋白质相互作用数据,甚至能够在全基因组范围内对蛋白质相互作用进行分析。然而,由于实验技术的限制,很多高通量实验方法测得的蛋白质相互作用数据的错误率都比较高。此外,传统实验的方法不适用于检测大规模数据。
针对这一科学问题,中国科学院新疆理化技术研究所多语种信息技术研究室硕士生王延斌在研究员尤著宏的指导下,经过系统的研究,提出了一种使用蛋白质序列信息预测蛋白质相互作用的计算方法。为了获得重要的蛋白质信息,科研人员首先使用位置打分矩阵(PSSM)去表示每一个蛋白质序列。研究发现打分矩阵的表示方法不仅保留了序列的位置信息,还保留了蛋白质的化学信息。同时,为了开发PCVMZM预测模型,科研人员首先在不同尺度的PSSM打分矩阵上提取到准确的、有代表性的蛋白质信息,并将每一个信息表示成一个特征向量作为特征,运用一个强分类器去预测蛋白质的交互。研究结果表明此方法能够提供精确、稳定、覆盖率高的预测信息,为基因组学研究提供了一个有用的决策工具。该研究成果发表在《国际分子科学杂志》(International Journal of Molecular Sciences)上,发表至今约两个月时间,被引用23次。
基于上述研究成果,科研人员通过构建一个深度学习系统,实现了更加准确、稳定的预测系统。实验结果表明使用了深度学习方法后,预测准确率可以提升了2.2%,并且可以实现跨物种检测。该研究成果发表在《生物分子》(Molecular Biosystems)上。
该工作得到国家自然科学基金、中科院的资助。
论文链接1,2
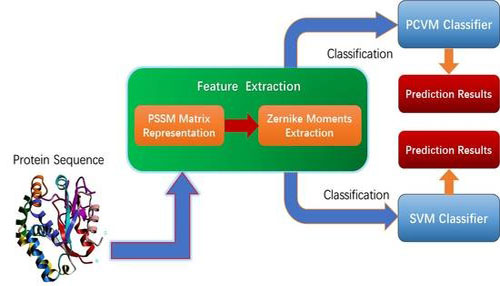
PCVMZM模型的工作流程
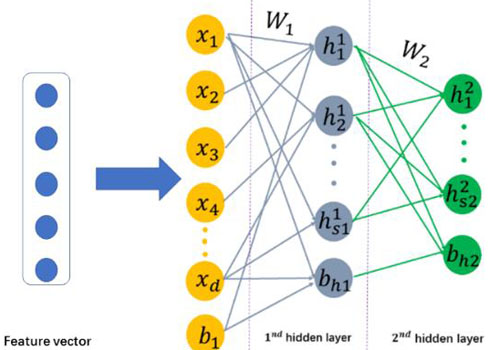
稀疏的深度结构




