中国科学院新疆理化所在构建大规模异构生物分子关联网络及其链路预测研究中取得进展
中国科学院新疆理化技术研究所多语种信息技术研究室研究生郭镇豪、易海成在尤著宏研究员的指导下,开展的关于大规模异构生物分子关联网络的研究“Construction and Comprehensive Analysis of a Molecular Association Network via lncRNA-miRNA-Disease-Drug-Protein Graph”(构建及综合分析基于长非编码RNA-微小RNA-环状RNA-信使RNA-微生物-疾病-药物-蛋白质的生物分子关联网络)于近日发表在《Cells》杂志上。
该工作在国际学术界首次提出了生物分子关联网络(Molecular Association Network,MAN)的定义,MAN网络系统的集成了8种不同类型的生物分子(包括lncRNA、miRNA、circRNA、mRNA、microbe、disease、drug及protein)及18种关联关系(包括miRNA-disease、circRNA-disease、circRNA-miRNA、disease-mRNA、disease-microbe、drug-disease、drug-mRNA、drug-microbe、drug-protein、lncRNA-disease、lncRNA-mRNA、lncRNA-miRNA、lncRNA-protein、miRNA-drug、miRNA-mRNA、miRNA-protein、mRNA-protein、protein-protein),并在此基础上建立了一个预测、发现生物分子间潜在关联关系的计算模型。多年来,不同生物分子之间如何相互作用以产生适当的细胞行为一直是系统生物学和基因组学界密切关注的科学问题。本工作一方面为整合大规模生物数据以构建快速、高效的计算模型提供了新思路,另一方面对促进理解基因的调控,揭示疾病的分子机理、加速药物靶标的发现以及药物重新利用具有重大意义。
后基因组时代的一个关键问题是如何系统地建模及表征细胞内转录物或翻译物之间的关联关系。高通量“组学”技术的快速发展为此任务奠定了数据基础,然而很多手工方法鉴定的相互作用的假阳性比较高并且耗时耗力,因此迫切需要开发计算机计算预测工具来为湿实验提供筛选和指引。针对这一科学问题,该工作依托于目前已有的多种孤立的生物分子关联关系,整合构建了一个涵盖长非编码RNA、微小RNA、环状RNA、信使RNA、微生物、疾病、药物及蛋白质的异构生物分子关联网络。在系统论和整体论的视角上,提出了一个基于节点属性和节点行为特征进行生物分子间潜在关联关系预测的计算框架。具体来说,节点的属性如蛋白质、RNA的序列通过k-mer特征来描述,疾病由其表征的语义相似性来表征,而药物由其化学结构的分子指纹来建模,节点的行为特征则通过图/网络嵌入学习模型LINE进行表示学习。每个节点通过结合上述属性和行为信息,进而被表征成一个低维、稠密的向量。最后通过机器学习模型随机森林来执行潜在关联关系预测任务。研究结果表明,此方法能够提供精确、稳定、覆盖率高的分子间关联关系预测结果。该工作为理解生物分子之间的协同关联关系提供了系统性的视角,将有助于启发和推动后续系统生物学及生物医学研究。《Cells》期刊评审人对这一重大突破给予了很高评价:“作者在展示如何将尽可能多的数据集成到学习框架中以预测新颖有意义的关联关系方面做出了得体的工作(decent work)。所呈现的结果是任何应用领域机器学习论文的典型结果。”
该工作得到国家自然科学基金优秀青年科学基金等资助。
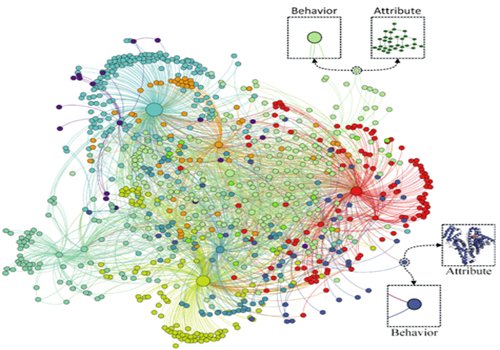
图 1.生物分子关联网络示意图
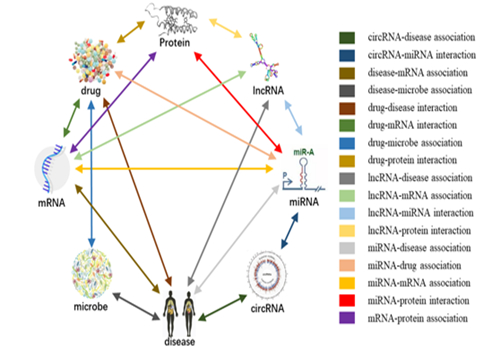
图 2.人类大规模异构生物分子关联网络中分子关联关系类型






